Using molecular dynamics to produce an ensemble of protein conformations for more biologically-relevant docking experiments

Ryuichiro Hara†, Stuart Firth-Clarke†, Nathan Kidley†
†Cresset, New Cambridge House, Bassingbourn Road, Litlington, Cambridgeshire, SG8 0SS, UK
Abstract
Docking is a method that utilizes protein structures to generate ligand binding poses and scores the interactions that are made. This method is used extensively in drug discovery to screen molecule designs and help prioritize them. Docking software typically treats the protein as a rigid body, whilst the ligand is conformationally flexible. Occasionally this is problematic because the protein structure is not in the biologically relevant conformation for a given ligand, which leads to the docking results not being relevant. In this case study, we will firstly validate the approach of using molecular dynamics (MD) to produce an ensemble of protein conformations to be used with ensemble docking in Flare.
Introduction
There are several reasons why an X-ray protein structure may not be in the biologically relevant conformation, for example, crystal packing artifacts from interactions within the crystal lattice, cofactors used in the purification or crystallization process, or if the protein is solved without the presence of a ligand in the active site. Furthermore, crystal structures represent a snapshot in time which does not reflect the dynamic nature of proteins. When docking treats the protein as a rigid body the lack of accounting for protein flexibility can lead to misleading results. Protein flexibility can mean a minor movement of an amino acid side chain to larger changes in the protein structure. An example of a moderate change in protein conformation that affects rigid docking comes from kinases, where the DFG-loop can be observed in at least two conformations, the DFG-In and DFG-Out conformation depending on the inhibitor bound. Not accounting for this change produces docking results that do not relate to the activity for all compounds.
Cambell et al.1 reported how they used molecular dynamics to sample protein flexibility and clustered the trajectory to generate an ensemble of protein conformations. They then docked ligands into the ensemble of protein conformations, treating each protein conformation rigidly, to indirectly incorporate protein flexibility into the docking protocol. Self-docking and cross-docking experiments were performed on examples from cyclin-dependent kinase 2 and factor Xa. This approach was developed to identify the lowest scoring pose with the correct binding mode for a given ligand.
A CDK2 and Factor Xa datasets2 were used to validate our approach. In both examples the X-ray protein structures were prepared, and dynamics simulations performed in Flare. The simulation trajectories were clustered to produce an ensemble of distinct protein conformations, which were then used in ensemble docking4 in Flare. Clustering of dynamics trajectories is a new feature that will soon be available in Flare. This workflow identifies the best protein conformation for docking each ligand, enabling the inclusion of protein flexibility in the docking workflow.
Flare is a user-friendly interface that also includes a Python API, enabling users to programmatically run or automate a workflow with an implicit distributed computing feature via the Cresset Engine Broker™. In our prototype workflow we developed Python scripts to accomplish this study.
Method
Protein preparation
All protein structures were prepared in Flare using the protein preparation tools. Structures were downloaded and checked for alternative conformations of amino acid side chains, choosing the preferred side chain rotamer either using the occupancy of the side chain or based on the contacts the side chain made. They were prepared using Flare’s automated protein preparation tool. This workflow adds hydrogen atoms, explores tautomer states of ionizable residues; can add missing sidechains, and fill gaps of two amino acids in the structure, caps terminal residues and optimizes residue side chains to maximize their interactions and the internal H-bond network. In most cases, the automatic protein prep in Flare is appropriately set-up for docking, energetic and molecular dynamic studies, but careful inspection is still recommended.
Molecular dynamics
Crystallographic water molecules around the ligand were kept and used in the Flare MD simulation. Most of the MD parameters were kept to the Flare defaults, in which the small molecule was charged by AM1-BCC and parametrized by OpenFF 2.0. Proteins were charged and parameterized by amber14ffsb. The hydrogen-mass repartitioning (HMR) was used with 4fs time-steps. The simulation was carried out for 4 ns after 100ps equilibration. This resulted in 2000 frames (2ps per frame) in every trajectory. Using the default setup, the user can run MD in a few clicks in within the Flare application, and a 4 ns simulation completes in less than an hour on a PC with a single GPU.
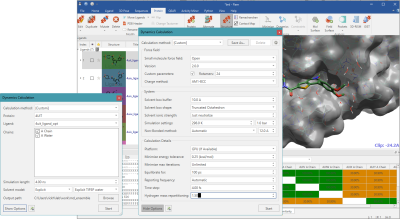
Figure 1: Molecular Dynamics setup in Flare.
Clustering of trajectories
The active site was defined as a set of residues within 6 Å of distance from the ligand in the co-crystal structure. The coordinate vector of the heavy atoms of the active site residue was used in the mass-weighted average linkage RMSD-based clustering algorithm1,2. Cambell et al. mention that 6-8 clusters are sufficient to make an ensemble, but here we took 20 clusters for sampling 20 representative conformations from the trajectory. Clustering may alternatively be done using the cpptraj function, which is provided as part of Amber tools, but we chose to implement clustering in Python for this preliminary experiment to explore different options and optimize the clustering results.
Time-dependent transitions between clusters were depicted in Figure 2 and fluctuation of heavy atoms in the active site was tracked by RMSD. The coloring in the plots in Figure 2 is by cluster membership, the size of the graph node corresponds to the frequency, and the length of the directed edge corresponds to the frequency of the transition (shorter more frequent). This shows the loop and progression of the protein structure space in the dynamics (though extremely frequent self-loops are omitted for clarity). Though the average linkage does not divide the size of a cluster evenly, RMSD-based variance within every cluster is calculated to identify the medoid regardless of the size of the cluster. These medoid conformations are all certain snapshots from the trajectory, thus should preferably be relaxed (minimized) using the same force field as for the MD process. As we are interested in only the immediate environment around the active site, atoms within a distance of 8 Å from the ligand were minimized for every snapshot structure.
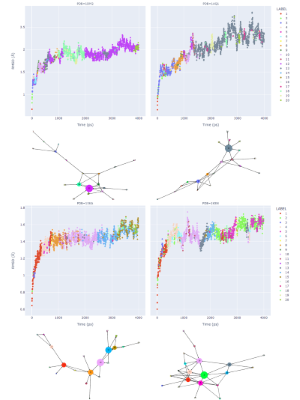
Figure 2: Analysis of dynamics trajectories, for each protein structure 1DM2, 1AQ1, 1XKA, 1KSN showing the RMSD values compared to the starting coordinates. The trajectories were then clustered, the results are shown in a directed graph. The coloring is consistent between each example for cluster nodes and cluster membership. The node size corresponds to the number of members in a cluster, and the length of an edge corresponds to the frequency of the transition (shorter more frequent). This shows the loop and progression of the protein structure space in the dynamics (though extremely frequent self-loops are omitted for clarity).
Docking calculations
The Lead Finder™4 algorithm built into Flare was used for all docking calculations. Docking runs for each chosen protein conformation (the cluster medoids and reference Xray structure) were iterated through the set of ligands. For every docking process, possible ligand poses were generated with various scores indicating the quality of fit to the binding site. While Lead Finder provides different scaled energy-based scoring (dG, VSscore and Rank Score), we used Rank Score which has been optimized is to identify the crystallographic ligand pose and dG score to compare the estimated binding poses of a ligand in the self and cross docking experiments.
The Flare ensemble docking workflow allows you to dock into multiple conformations of a protein. We used the Flare Python API to write pyflare scripts to execute the docking experiments, tuned all settings for the experiments that are available in GUI, such as adding unified constraints to all proteins, and wrote out the results in a custom format. All high-scoring poses were written to an SDF file across the ensemble + X-ray protein. This enables the user to quickly advance to analysis using other external tools such as Jupyter notebook.
The protocol followed in this work is summarized in Figure 3. The steps highlighted in yellow were executed within Flare; the clustering process which was written in Python and executed by the pyflare API.
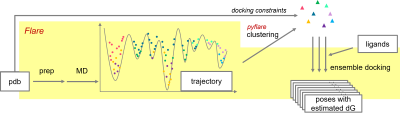
Figure 3: Workflow of MD/ensemble docking in Flare/pyflare.
Results
Verification of MD ensemble docking
As explained in Cambell’s paper, we verified the protocol by self-docking and cross-docking of two ligand sets: two CDK2 ligands from PDB structures 1DM2 and 1AQ1 (Figure 4 – left); and two Factor Xa ligands from PDB structures 1XKA and 1KSN (Figure 4 – right). The CDK2 ligands provide an example of a large size difference in the bound ligands. The Factor Xa ligands instead are highly flexible. In both these cases, cross-docking may often fail.
For the CDK2 example (Figure 4 – left), both the HMD crystallographic ligand (from 1DM2.pdb) and STU (from 1AQ1.pdb) self-docked into their respective X-ray protein structure within 1Å RMSD. Cross-docking of the HMD ligand into 1AQ1.pdb was reasonably successful with a RMSD = 1.554 Å. However, due to the large size of the ligand and the resulting differences in the pocket size, cross-docking of STU to 1DM2.pdb failed to produce a correct solution.
Self-docking experiments for both the CDK2 (HMD and STU) against the ensemble of MD snapshots from their respective protein structures also gave good results, as would be expected. Cross-docking experiments using the ensemble of MD snapshots for the cross-docking experiments was successful, unlike the previous direct cross-docking experiment. The results produced docked poses with RMSD <2 Å (STU 1.255 Å and HMD 1.654 Å) when compared to the original X-ray ligand. It is worth noting that in addition to such acceptable RMSD requirements, the docked poses were also examined visually to confirm that they are similar to the crystallographic poses.
For the more difficult case of Factor Xa with highly flexible ligands, self-docking of ligands 4PP (from 1XKA.pdb) and FXV (from 1KSN.pdb) into their native protein structures was successful (Figure 4 – right). Cross-docking experiments for Factor Xa were not as successful, producing poses with a RMSD >2 Å for both ligands with poor docking scores. Visual inspection of these poses confirmed the results did not identify crystallographic pose of either ligand.
Self-docking experiments for both the 4PP and FXV ligands against the ensemble of MD snapshots from their respective protein structures generated slightly improved RMSD values with respect to rigid docking to crystallographic proteins. Using the ensemble of MD snapshots for cross-docking Factor Xa ligands produced poses within an acceptable range (RMSD FXV 1.385 and 4PP 1.498) with an LF dG scores close to the values observed in the self-docking experiments.
.png)
Figure 4: Verification of MD/ensemble docking. Left: self-docking and cross-docking results for CDK2 ligands. Right: self-docking and cross-docking results for Factor Xa ligands.
Overall, these results from both the CDK2 and Factor Xa examples demonstrate that the MD-clustering-ensemble docking method in Flare can successfully produce ensembles of protein conformations which enable to incorporate protein flexibility in a docking workflow. In these examples the differences in the active sites are mainly observed as side chain movements of the active site, and as such the relatively short 4ns MD simulations used is sufficient to sample these changes. Larger changes in the protein conformation would likely require longer MD simulations to generate a suitable ensemble of conformations for ensemble docking.
Conclusions
In this work, we presented an approach that makes use of the ensemble docking workflow in Flare together with molecular dynamics to perform docking experiments, while taking into consideration protein flexibility. This is particularly useful as an approach when the available protein structure is not in the biologically relevant conformation for your ligand, which could be manifested as amino acid side chain movements or a larger change.
The molecular dynamics clustering method used here is being developed for implementation in the upcoming release of Flare. It is a useful analysis method for MD simulations as it enables the generation of different protein conformations, in this example to take protein flexibility into account for docking experiments.
In this work, Python scripts were written to automate the entire process of protein preparation, ensemble docking and results analysis. These scripts where saved and executed from a Jupyter notebook.
References
- A.J. Cambell et al., J. Chem. Inf. Model. (2013)
- J. Shao et al., J. Chem. Theory Comput. (2007)
- Lead Finder, 2112 build 1, BioMolTech®, Toronto, Ontario, Canada























